





















In a previous blog, we shared how Paragon™ Pathfinder plays an important role in closed-loop automation by tuning the paths of RSVP or Segment-Routed Traffic Engineered LSPs according to changing conditions that it observes in the live network. This blog will explore another example which ensures that latency-sensitive traffic always follows the minimum latency path across the network, even when the latency conditions in the network change. Such LSPs can be used to underpin latency-sensitive classes of traffic in business VPN deployments or to underpin Ultra-Reliable Low Latency Communications (URLLC) traffic in 5G deployments.
Key Components
Paragon Pathfinder uses periodic measurements of the latency between directly connected nodes in the network. These periodic measurements are carried out by the TWAMP-Light protocol. This involves each node sending time-stamped probe packets to its neighbors in order to determine the current latency of each of its links. In the WAN, the latency is strongly dominated by the time-of-flight through the optical fiber, which is approximately 5 microseconds per kilometer. When compared to this, the latency within the router is normally negligible, typically of order a few microseconds. If a router interface is momentarily congested due to a microburst causing the offered load to be greater than the bandwidth of the link, then the extra latency incurred could rise to tens of milliseconds, depending on the depth of the buffer. Of course, if the buffer overflows due to sustained over-subscription of the link, some packets are dropped. However, if Pathfinder’s automated congestion avoidance is being used, such congestion should not occur.
The link latency information measured by TWAMP-Light is flooded via the IGP protocol throughout the domain. Extensions have been defined in the IGPs to carry this delay-metric information. It’s a best practice to configure a threshold for the amount of latency change required to trigger an update to the IGP delay-metric. This helps to avoid an excessive volume of IGP updates triggered by minor fluctuations in latency.
Now, let’s see how the latency information reaches Pathfinder.
Pathfinder has BGP-LS sessions with one or more routers in the domain. The BGP-LS speakers, when communicating the topology of the network to Pathfinder, also communicate the delay metric of each link. If the latency of a link changes, a BGP-LS message is automatically sent to Pathfinder, so it knows about the change. In this way, Pathfinder can see the topology of the network and the latency of each link, as shown in Figure 1.
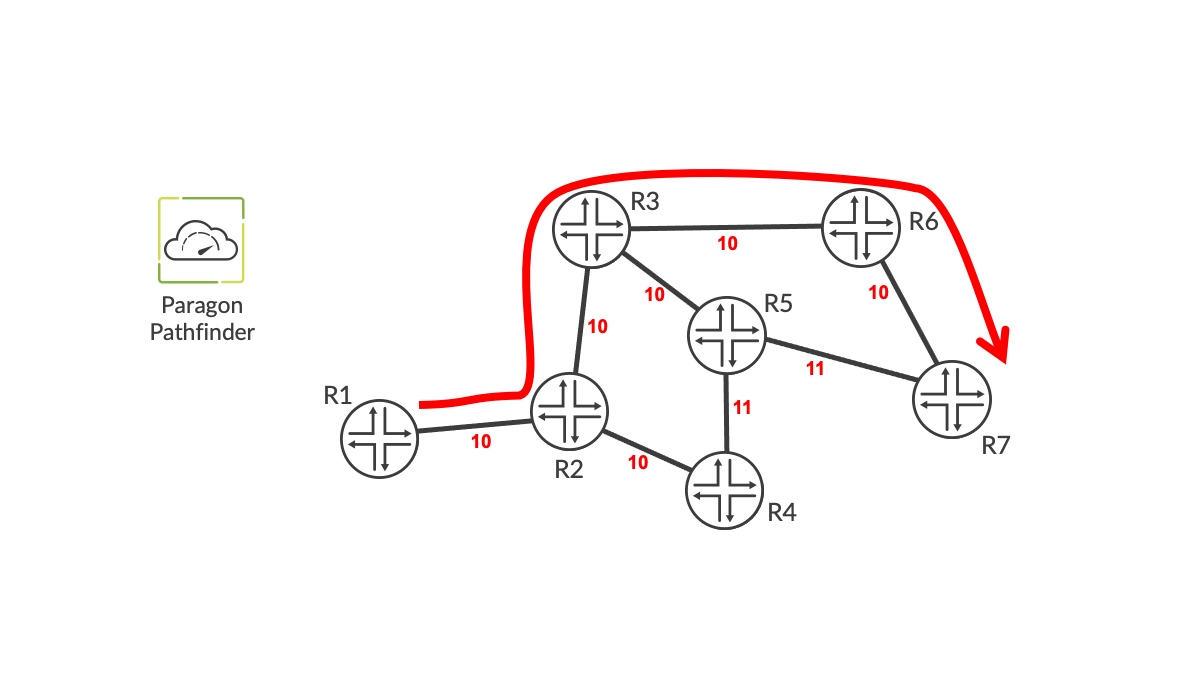
Creating Minimum Latency Paths
Suppose we want an LSP to run from R1 to R7 that has the minimum possible latency between the endpoints. Pathfinder computes the path using delay as the cost function and then instantiates the LSP on R1 via the PCEP protocol. This results in the path shown in Figure 2 where the total latency end-to-end is 40 units (the minimum possible given the delay values on the various links).
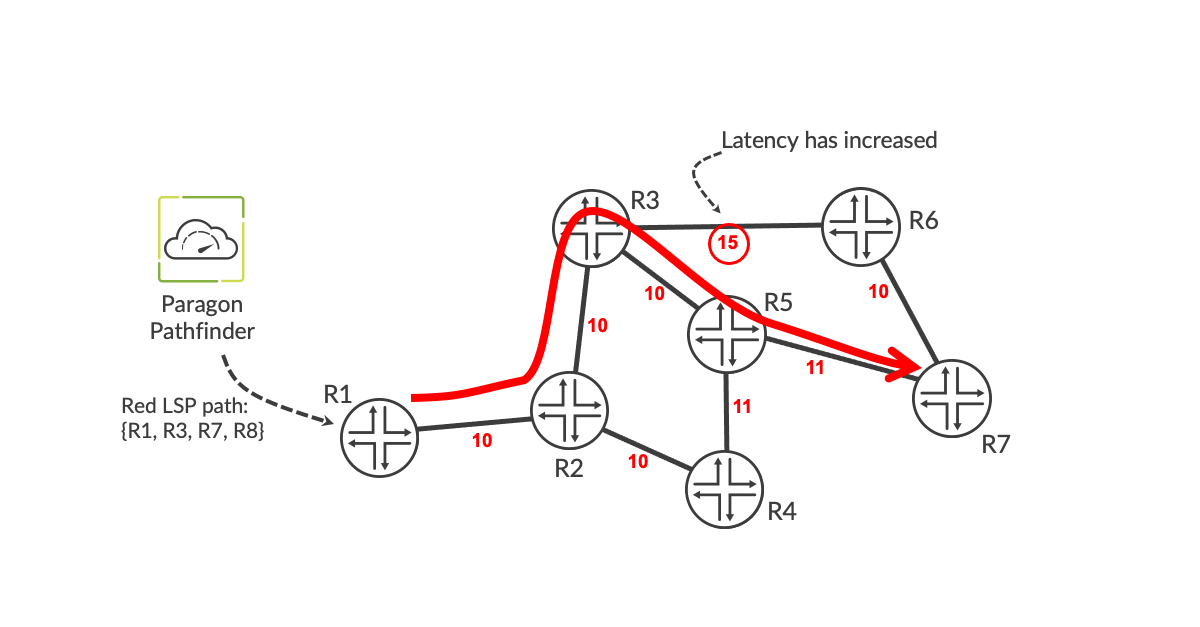
However, the latency conditions can change over time. This usually occurs when there is a change in the underlying L1 or L2 connectivity between routers. Such a change can be be due to a fiber cut. When the fiber is cut, optical protection/restoration kicks in, often resulting in a longer path through the underlying optical layer. Alternatively, if the operator of the network shown is buying bandwidth from a wholesaler that is implementing the connectivity as a carrier ethernet service or MPLS pseudowire, the latency could change due to underlying rerouting within the wholesaler’s network. Although the precise cause of such latency changes may not be known to the operator of the network shown in Figure 1, the increase in latency is nevertheless detected and can be acted upon by Pathfinder. In Figure 2, the latency of the link between R3 and R6 has increased from 10 to 15 units. Pathfinder recomputes the path of the low-latency LSPs that are using the link, in case the current paths are no longer optimum given the increase in latency on the link. In the example, Pathfinder sees that the lowest latency path is now R1-R3-R5-R7. It sends a PCEP message containing the details of the new path to the ingress router, which moves it accordingly in a make-before-break manner, so no traffic is lost during the path change.
In addition to automatically tuning the paths of low-latency LSPs, Pathfinder stores latency information in a time-series database. This enables Pathfinder to generate charts showing the delay as a function of time on a given link or a given LSP. This is useful for troubleshooting, if customers/end-users have noticed issues with excessive delay at the application level.
A previous blog shared how Pathfinder automatically diverts the paths of some LSPs that are using congested links in order to ease congestion in the network. However, because the diverted path is likely to be longer, it’s best to avoid diverting minimum-latency LSPs. Instead, the LSPs that are carrying non-latency-sensitive traffic should be diverted. Pathfinder can be configured to behave in this way automatically.
In Summary
This blog explored another great example of closed-loop automation. Pathfinder can create minimum-latency LSPs, automatically tuning their paths on an ongoing basis, according to changing latency conditions in the network. This completely removes the manual labor associated with maintaining minimum-latency paths.


